После формулировки одночастичного самосогласованного подхода
для движения электрона в молекулярных и кристаллических структурах основой
молодой науки
квантовой химии, а также физики твердого тела стало нахождение решения
уравнения Шредингера для электрона, движущегося в самосогласованном потенциале
,
создаваемом остальными электронами и ядрами окружающих атомов:
|
Вначале, в соответствии с приближением, предложенным Хартри, данный потенциал определялся как сумма электростатических взаимодействий данного электрона со всеми другими электронами (отталкивание) и со всеми ядрами окружающих атомов (притяжение):
|
где
суммарная
электронная и ядерная плотность в точке
.
К сожалению, в этом приближении полностью игнорировались корреляционные
эффекты, а также чисто квантовое свойство неразличимости частиц-электронов,
являющихся фермионами. Известно же, что в соответствии с законами квантовой
механики неразличимые частицы при их электростатическом взаимодействии могут к
тому же еще меняться местами. При этом в матричных элементах дополнительно
появляется общий множитель, равный
1
для фермионов (фермионная петля). Из-за этого множителя между электронами
возникает дополнительное притяжение, полностью игнорируемое в приближении
Хартри. Это игнорирование обменного взаимодействия часто приводило к большим
погрешностям в определении свойств объекта, особенно в случае небольших
молекул.
Этот недостаток был устранен (частично) в приближении Хартри
Фока,
где обменный член был явно включен в уравнения Хартри
Фока
(см. (1.22)). К сожалению, уравнения Хартри
Фока
перестали быть линейными уравнениями относительно
.
В них обменное взаимодействие с другим электроном приводило к перемещению
электрона в точку
.
Это стало очень большой проблемой при решении данных нелинейных уравнений при
их разложении по любым базисным функциям. Имеющиеся мощные алгоритмы диагонализации
систем линейных уравнений, применимые в приближении Хартри, тут оказались
невостребованными. Эти вычислительные проблемы сказываются для приближения
Хартри
Фока
в том, что данный метод является очень медленным, он применяется обычно только
для небольших молекул. Вычислительная шкалируемость (количество числовых
операций) для него обычно составляет N4, где N
число
базисных функций (или атомов).
Из-за данных проблем появилась необходимость в подходе, где
квантовое свойство обмена (а также корреляционные эффекты) вычислялось бы
иначе, не приводя к нелинейным уравнениям типа Хартри
Фока.
Первой теорией, показавшей путь решения данной проблемы, явилась теория Томаса
Ферми,
впервые предложенная в [13]. Хотя в ней не учитывался обмен, но, тем не менее,
был показан метод нахождения кинетического вклада в гамильтониан без явного
решения уравнения Шредингера.
Согласно теории Томаса
Ферми
(ТФ), любая молекулярная или кристаллическая структура представлялась в виде
электронной жидкости с включенными в нее ядрами атомов. При этом считается, что
электроны являются полностью делокализованными, т. е. их волновые функции
являются комбинацией плоских волн. Также постулируется принцип, что все
уравнения, применимые для случая однородного распределения, т. е. случая,
когда электроны движутся на фоне однородно размазанного положительного заряда
(модель желе), остаются справедливыми и для случая, когда однородный
положительный фон заменяется потенциалами ядер в виде δ-функций. Для
однородного распределения и для обеспечения принципа минимума энергии при
выполнении принципа Паули электроны в импульсном пространстве должны заполнить
все состояния, вплоть до квазиимпульса Ферми kF. Плотность однородного электронного газа
имеет следующий вид [14]:
|
(1.49) |
Плотность кинетической энергии системы невзаимодействующих электронов можно получить из следующего выражения:
|
где
электронная
плотность.
Таким образом, кинетическая энергия в теории ТФ может быть заменена:
|
где
[15].
Функционал энергии, связанный с межэлектронным взаимодействием, т. е. вклад электростатического кулоновского взаимодействия (вклад Хартри), будет иметь следующий вид:
|
Пусть теперь взаимодействующие электроны движутся в
некотором внешнем потенциале
.
Энергия, связанная с внешним потенциалом, может быть записана следующим
образом:
|
Минимизируем функционал полной энергии системы E[ρ] при условии, что число электронов сохраняется:
|
Используя метод неопределенных множителей Лагранжа, требуем, чтобы выражение
|
(1.55) |
имело минимум. Таким образом,
получаем известное уравнение Томаса
Ферми
[13, 16]:
|
где λ можно ассоциировать с химическим потенциалом системы.
Ранее теория ТФ достаточно широко использовалась, так,
например, она была подробно рассмотрена в обзорах [17, 18]. Также в работах [19, 20] были изучены некоторые аспекты данного приближения.
В частности, там было показано, что теория Томаса
Ферми
является точной в случае предела бесконечных зарядов ядер.
Введем следующие обозначения:
|
|
|
и, выразив из (1.56)
,
получим
|
Таким образом, между электронной плотностью и внешним потенциалом существует взаимно однозначное неявное соотношение.
Теория Томаса
Ферми
с достаточной точностью описывает некоторые качественные характеристики, такие
как полные энергии атомов, но в вопросах химии и материаловедения, включающих
рассмотрение валентных электронов, она практически бесполезна, например, из нее
не следует наличие химической связи.
Важно то, что выражение (1.59) сформулировано в терминах электронной плотности, а не
волновых функций, как в уравнениях теории Хартри
Фока.
Теория Томаса
Ферми,
несмотря на свою простоту и явные дефекты, формулирует крайне важный вывод:
электронная плотность
может полностью характеризовать систему.
Действительно, если подставить
в (1.59), то можно получить
,
а из (1.58)
.
Кроме того, проинтегрировав плотность
по объему, получим полное число электронов. Таким
образом, физическая система полностью характеризуется заданной электронной
плотностью
[21].
ТФ-теория подтолкнула Кона (Kohn) и Хоэнберга (Hohenberg) к разработке теории
функционала плотности (ТФП)
Density Functional Theory (DFT) [22], за которую впоследствии Кон был удостоен
Нобелевской премии по химии в 1998 г.
Суть работы состоит в том, что все свойства электронной структуры системы в невырожденном основном состоянии полностью определяются ее электронной плотностью:
|
где сумма идет по N низшим занятым состояниям,
волновая
функция электрона.
Как известно, система N электронов (в адиабатическом приближении[2]) описывается следующим гамильтонианом:
|
Основное состояние энергии определяется путем минимизации функционала энергии E[ψ]:
|
Член
и количество электронов N полностью определяют все свойства
системы для основного состояния (в дальнейшем будет рассмотрено только такое состояние).
Количество электронов можно определить из условия (1.54).
Первое положение теоремы Хоэнберга и Кона состоит в том, что
плотность
основного состояния связанной системы
взаимодействующих электронов в некотором внешнем потенциале
однозначно определяет этот потенциал [23].
Замечания:
1. Термин «однозначно» здесь означает «с точностью до не представляющей интереса аддитивной константы».
2. В случае
вырожденного основного состояния положение относится к плотности
любого основного состояния.
3. Положение является математически строгим.
Доказательство теоремы:
Пусть
плотность
невырожденного основного состояния системы N электронов в потенциале
,
отвечающего основному состоянию Ψ1 и энергии E1.
Тогда
|
где H1
полный
гамильтониан, соответствующий
,
а T и Uee
операторы
кинетической энергии электронов и энергии межэлектронного взаимодействия.
Теперь допустим, что существует второй потенциал
,
и ему отвечает основное состояние
,
приводящее к той же самой плотности
.
Тогда
|
Поскольку состояние Ψ1 предполагается невырожденным, принцип минимума Рэлея
Ритца
для Ψ1 приводит к неравенству
|
Аналогично
|
где использован знак нестрогого неравенства ≤, поскольку невырожденность Ψ2 не предполагается. Сложение неравенств (1.65) и (1.66) приводит к противоречию:
|
(1.67) |
Следовательно, согласно доказательству от противного,
предположение о существовании второго потенциала
,
который не равен
,
но дает ту же самую плотность
,
не может быть верным. Таким образом, для невырожденного состояния положение
доказано.
Поскольку плотность
определяет как число частиц N, так и потенциал
(с точностью до несущественной аддитивной
постоянной), с помощью нее можно получить полный гамильтониан H и оператор числа частиц N для электронной системы. Следовательно,
неявно определяет все свойства, получаемые
из H путем решения зависящего
или не зависящего от времени уравнения Шредингера (даже при наличии дополнительных
возмущений типа воздействия электромагнитного поля).
Замечания
1. Требование невырожденности легко снимается [24].
2. Положение, естественно, остается справедливым и в особом случае невзаимодействующих электронов [21].
Таким образом, функционал полной энергии может быть записан в виде
|
(1.68) |
Группируя первый и второй члены в (1.68), получим
|
(1.69) |
Здесь FHK
универсальный
функционал Хоэнберга
Кона,
вид которого не зависит ни от числа частиц, ни от внешнего потенциала.
Известно, что для системы N частиц функционал энергии
|
имеет минимум при
,
где Ψ
волновая
функция основного состояния системы. Пусть
характеризует основное состояние системы с
другим внешним потенциалом. Тогда из (1.70) и (1.68) ясно, что
|
Если с помощью интегрирования некоторой электронной
плотности
может быть получено определенное количество
электронов в системе, следовательно, полная энергия, рассчитанная с этой
плотностью, будет не меньше точного значения энергии системы в основном
состоянии.
Минимизируем функционал энергии (1.69). Воспользуемся, так же как и в методе ХФ и ТФ, методом неопределенных множителей Лагранжа. Учитывая условие (1.54), запишем
|
где μ
неопределенный
множитель Лагранжа, который можно ассоциировать с химическим потенциалом
системы
|
Если известна точная форма функционала FHK, решение уравнения (1.73) будет точным для электронной плотности, находящейся в основном состоянии. Однако, к сожалению, точное выражение для этого функционала получить очень сложно.
Рассмотрим случай невзаимодействующих электронов, тогда FHK[ρ] = T[ρ], и (1.69) переходит в следующее выражение:
|
Известно, что в случае невзаимодействующих электронов можно
найти энергию и плотность основного состояния, вычислив собственные функции
и собственные значения εm следующих одночастичных
уравнений без взаимодействия:
|
В результате получаем
|
Таким образом, энергию основного состояния можно представить так:
|
Рассмотрим теперь случай взаимодействующих электронов.
Функционал Хоэнберга
Кона
в этом случае будет иметь вид
|
где
функционал
кинетической энергии невзаимодействующих электронов, а
функционал
энергии межэлектронного взаимодействия Хартри:
|
Последнее слагаемое
так
называемый функционал обменно-корреляционной энергии, и формула (1.78) является для него определением.
Выражение (1.72) в этом случае имеет вид
|
Перепишем это уравнение в следующем виде:
|
(1.81) |
где
|
Здесь
|
Таким образом, получаем следующее уравнение:
|
и
|
Уравнение (1.84) по виду идентично (1.75) для невзаимодействующих частиц, движущихся в некотором
эффективном внешнем потенциале
вместо
,
и называется уравнением Кона
Шэма
[25], а ψm
спин-орбиталями
Кона
Шэма.
Подставив (1.82) в (1.69), получим
|
Принимая во внимание (1.85), получим следующее выражение для энергии основного состояния [26]:
|
(1.87) |
Обобщая вышесказанное, необходимо отметить, что:
Ø Многоэлектронная система рассматривается как система невзаимодействующих электронов, в которой каждый электрон движется в эффективном потенциале, создаваемом другими электронами.
Ø Физический смысл имеет только минимум функционала E[ρ], связанный с основным состоянием системы.
Как уже было показано в (1.83), обменно-корреляционный потенциал есть вариационная производная обменно-корреляционного потенциала по электронной плотности. Для однородного электронного газа он зависит от электронной плотности. Для неоднородной системы значение обменно-корреляционного потенциала в точке, определяемой радиус-вектором r, зависит не только от значения электронной плотности в этой точке, но и от ее вариации по координате вблизи этой точки. Следовательно, обменно-корреляционный потенциал можно записать в следующем виде:
|
(1.88) |
В приближении локальной плотности (ПЛП)
Local Density Approximation
значение
обменно-корреляционной энергии равно известному значению энергии многоэлектронного
взаимодействия в электронной системе с постоянной электронной плотностью
(однородный электронный газ). Обменно-корреляционная энергия в этом случае
равна
|
(1.89) |
|
(1.90) |
где
|
(1.91) |
Здесь
обменно-корреляционная
энергия однородного электронного газа с плотностью ρ [22].
Данная энергия легко может быть вычислена методом, схожим с методом ТФ, в
предположении знания волновых функций в виде плоских волн, заполняющих все состояния,
вплоть до kF.
Выражение для обменного вклада будет иметь следующий вид:
|
(1.92) |
где
|
(1.93) |
а rs
радиус
сферы, приходящейся на один электрон, определяется следующим образом:
|
(1.94) |
Впервые оценка корреляционного вклада была сделана Е. П. Вигнером [27]:
|
(1.95) |
а позднее улучшена Д. М. Сиперли (Ceperly) [28, 29], который с помощью метода Монте-Карло вычислил его с высокой точностью (~1 %) [21]. В работе [30] была предложена следующая форма записи корреляционного вклада:
|
(1.96) |
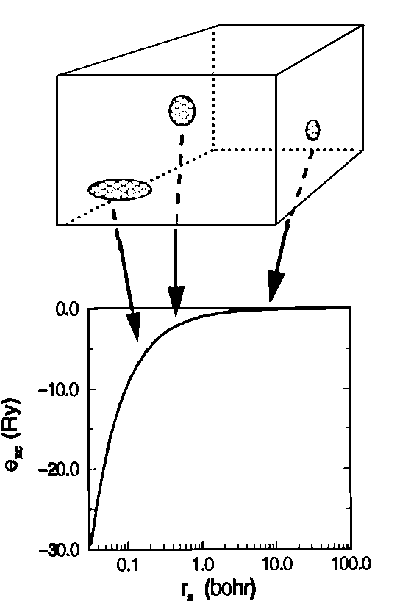
Рис. 1-4. Иллюстрация идеи приближения локальной плотности. Распределение электронного газа в некотором бесконечно малом объеме можно считать однородным, таким образом, его вклад в обменно-корреляционную энергию по величине равен вкладу однородного электронного газа такого же объема. Ось абсцисс пропорциональна радиусу сферы, приходящейся на один электрон. По оси ординат отложена обменно-корреляционная энергия однородного электронного газа [31].
Замечания:
1. Приближение локальной плотности с высокой точностью описывает типичные металлы, а также с достаточно хорошей точностью описывает системы с высокой электронной плотностью, например, переходные металлы [26].
2. Решение уравнений Кона
Шэма
в приближении локальной плотности лишь немногим более трудоемко, чем решение
уравнений Хартри, и гораздо проще, чем решение уравнений Хартри
Фока.
При этом типичная точность расчета обменной энергии КШ
порядка
10 %, в то же время обычно меньшая по величине корреляционная энергия
существенно завышается, как правило, примерно в 2 раза. В большинстве случаев
обе ошибки частично сокращаются.
3. Из практики расчетов известно, что ПЛП дает
энергии ионизации атомов, энергии диссоциации молекул и энергии связи твердых
тел с неплохой точностью, обычно 10
20 %.
При этом длины связей и, следовательно, геометрическое строение молекул и
твердых тел получаются в ПЛП, как правило, с очень хорошей точностью ~1
2 %.
4. Приближение локальной плотности, как и приближение спиновой плотности (обобщение ПЛП для систем с неспаренными спинами), может оказаться непригодным в некоторых случаях, например, для систем с тяжелыми фермионами, когда корреляционные эффекты электрон-электронного взаимодействия (для d-, f-электронов) столь сильны, что эти системы теряют всякое сходство с невзаимодействующим электронным газом [21].
В обобщенном градиентном приближении (ОГП)
Generalized Gradient Approximation
обменно-корреляционный
функционал зависит не только от плотности, но и от ее первой пространственной
производной [32]:
|
(1.97) |
Для расчета твердых тел наиболее широко используется приближение ОГП, предложенное группой Пердью (Perdew) [33]. Такое ОГП удачно исправляет многие дефекты ПЛП. Так, например, с помощью ОГП было верно определено основное состояние для ферромагнетика Fe и антиферромагнетиков Cr и Mn. Также были правильно предсказаны магнитообъемные и магнитоструктурные эффекты [34, 35]. Однако существуют случаи, где использование ОГП ведет к отсутствию связывания между атомами. Например, в случае димеров атомов благородных газов и молекулярного кристалла N2 использование ОГП приводит к абсурдному результату: отсутствию связи в данных структурах [36].
В заключение следует отметить, что описание обменно-корреляционных эффектов в ПЛП и в ОГП абсолютно непригодно для тех систем и подсистем, где начальное приближение невзаимодействующего электронного газа с медленно меняющейся плотностью ρ(r) принципиально некорректно. Примерами таких систем являются: электронный вигнеровский кристалл; системы с преобладанием ван-дер-ваальсовых (т. е. поляризационных) взаимодействий неперекрывающихся подсистем; системы, имеющие границы, где хвосты электронных плотностей стремятся к нулю в вакууме вблизи поверхностей [21].
Часто, особенно для случая расчета структуры атома, используются решения уравнений КШ в сферических координатах. В случае сферического потенциала V(r), учитывая разложение лапласиана
|
(1.98) |
где Λ действует на углы, составляемые вектором r с осями координат, получим
|
(1.99) |
где Ylm(r)
собственные
функции оператора момента импульса.
Используя (1.98), (1.99), можно видеть, что (1.84) переходит в следующее уравнение [37]:
|
(1.100) |
где второй
член
это
так называемый центробежный вклад, ответственный, в частности, за то, что волновая
функция электрона для
(p-, d-, f-оболочки) стремится к нулю при
.
На следующей блок-схеме (рис. 1-5) представлен алгоритм расчета с помощью метода теории функционала плотности.
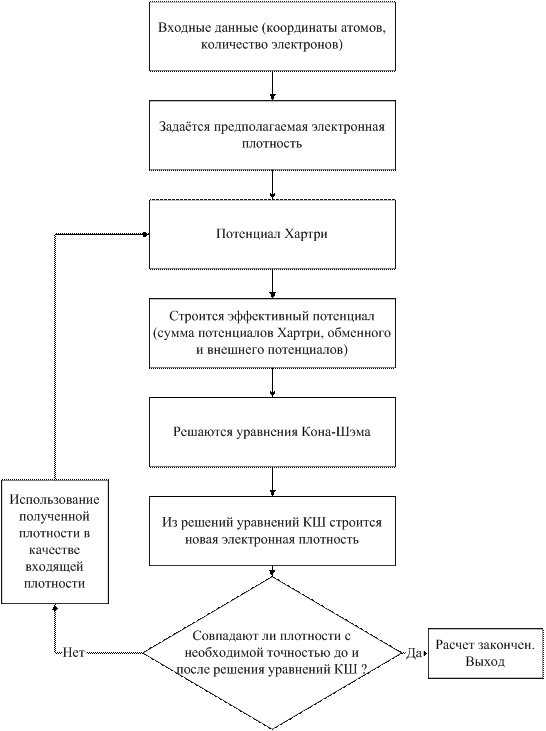
Рис. 1-5. Блок-схема алгоритма расчета с помощью теории функционала плотности.
Входные данные: В качестве входных данных задаются координаты атомов, заряды ядер, полное число электронов. В случае использования метода псевдопотенциалов задается также форма псевдопотенциала для всех различных типов атомов в системе. Если в качестве базисных функций используются плоские волны, необходимо проверять результаты расчета, поскольку они зависят от многих параметров: энергии обрезания, числа k-точек при интегрировании зоны Бриллюэна и др.
Предполагаемая электронная плотность: Задается первоначальная электронная плотность. Она может быть получена как сумма зарядов заданной первоначальной атомной конфигурации (нейтральные атомы и т. д.) или из предварительных полуэмпирических расчетов.
Построение потенциала Хартри: Построение потенциала Хартри может быть осуществлено с помощью решения уравнения Пуассона [39]
|
(1.101) |
Построение обменного потенциала: Для заданной электронной плотности строится VXC из (1.83).
Построение эффективного потенциала: Строится Veff из (1.82).
Проверка совпадения плотностей до и после решения уравнений КШ: Если интегральная разница между полученной плотностью и плотностью на предыдущей итерации по модулю больше, чем заданное изначально значение, процедура решения повторяется до сходимости.