![]()
![]()
![]()
Глава II.6.2.
Полуэмпирические
методы расчета электронной структуры (Semi-empirical)
Электронную структуру исследуемых молекул в программе HyperChem можно рассчитывать способами: используя полуэмпирические методы расчета, либо – неэмпирический метод Хартри-Фока, сделав выбор в меню Setup.
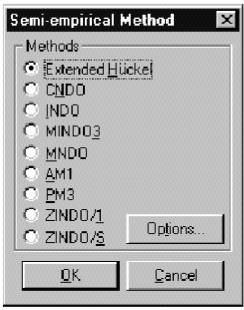
Полуэмпирические методы расчета можно использовать для всех типов расчетов в меню Compute. Полуэмпирические методы решают уравнение Шредингера для атомов и молекул с использованием определенных приближений и упрощений. Все методы этой группы характеризуются тем, что: расчет ведется только для валентных электронов; пренебрегаются интегралы определенных взаимодействий; используются стандартные не оптимизированные базисные функции электронных орбиталей и используются некоторые параметры, полученных в эксперименте. Экспериментальные параметры устраняют необходимость расчетов ряда величин и корректируют ошибочные результаты приближений. Необходимо помнить, что полуэмпирические методы в программе HyperChem могут обрабатывать не все элементы таблицы Менделеева, а только те, параметры которых внесены в файлы параметров.
Большинство доступных в программе HyperChem полуэмпирических методов включают схему для устранения вычислений, которые происходят со значительными затратами процессорного времени, в основном – расчета ряда интегралов перекрывания, а метод INDO (Intermediate Neglecting of Differential Overlap) (см. далее) не вычисляет и интегралы расталкивания, которые должны иметь небольшие величины.
HyperChem также позволяет Вам рассчитывать электронную структуру только части системы, используя смешанные методы вычисления. Например, можно изучить электронную структуру активного центра белка с использованием полуэмпирических методов расчета, учитывая оставшуюся часть белка и молекул растворителя в рамках метода молекулярной механики. Для этого, перед тем, как начинать расчет, выделите нужную часть системы с использованием инструментария меню Select, а затем, введите соответствующие параметры меню Setup и Compute. Необходимо подчеркнуть, что такие расчеты возможно проводить только в том случае, если выделенная часть системы не соединена формальными химическими связями с остальной частью молекулярной системы. (Например, построив модель белка можно удалить соответствующие химические связи активного центра, электронную структуру которого мы должны исследовать, а затем выделить активный центр с использованием различных способов меню Select. Например, выбрать параметр Molecules и выделить активный центр одним L-нажатием, либо выделить в нужной части один атом, а затем выбрать пункт Extend to sp3 в меню Select, при этом будет выделена вся молекулярная система, в которую входит выбранный атом. В этом случае программа HyperChem квантово-химически рассчитывает только выделенную часть атомов, а остальные рассматривает только как некий потенциал. В процессе оптимизации геометрии координаты не выделенной части атомов являются фиксированными и не изменяются в ходе проведения расчетов.
Расширенный
метод Хюккеля (Extended Huckel) (РМХ) предназначен для вычислений молекулярных
орбиталей и не позволяет оптимизировать геометрию и проводить
молекулярно-динамические расчеты. В нем используется приближение
невзаимодействующих электронов и в нем не используется приближение
самосогласованного поля (SCF)
Метод CNDO (Complete Neglect of Differential Overlap, полное пренебрежение дифференциальным перекрыванием) является простейшим методом SCF. Он используется для расчетов основного состояния электронных характеристик систем с открытой и закрытой оболочками, оптимизации геометрии и полной энергии.
Метод INDO (Intermediate Neglect of Differential Overlap, частичное пренебрежение дифференциальным перекрыванием) улучшает метод CNDO за счет учета расталкивания электронов на одном атомном центре. Позволяет проводить расчет основного состояния систем с открытой и закрытой оболочками, оптимизации геометрии и полной энергии. Это SCF метод.
Метод MINDO3 (Modified INDO, version 3, улучшенный метод INDO, версия 3) является дальнейшим развитием и расширением метода INDO. Для многих взаимодействий в нем используются эмпирические параметры вместо соответствующих вычислений. Этот метод позволяет получать хорошие результаты для больших органических молекул при расчетах основного состояния систем с открытой и закрытой оболочками, оптимизации геометрии и полной энергии. Это SCF метод.
Метод MNDO является дальнейшим развитием метода MINDO3, в котором исправлен ряд ошибок последнего. Позволяет проводить качественные расчеты электронной и атомной структур органических молекул, содержащих атомы 1-й и 2-й главных подгрупп (но не атомов переходных элементов). Этот метод позволяет получать хорошие результаты для больших органических молекул при расчетах электронных характеристик системы и теплот образования. Это SCF метод.
Метод AM1 является улучшением метода MNDO. Один из наиболее точных методов. Используется для органических молекул, содержащих элементы из главных подгрупп 1 и 2 групп периодической системы. Возможно, этот метод позволяет получать более качественные результаты, по сравнению с методом MNDO, для молекул, содержащих как азот, так и кислород. Вычисляет электронную структуру, оптимизирует геометрию, рассчитывает полную энергию и теплоты образования. Это метод SCF.
Метод PM3 является версией метода AM1. PM3 отличается от AM1 только величинами параметров. Параметры для PM3 были получены сравнением большого числа и вида экспериментов с результатами расчетов. Как правило, нековалентные взаимодействия в методе PM3 являются менее расталкивающими, нежели чем в AM1. PM3 первоначально предназначался для расчета для органических молекул, но потом он был также параметризован и для ряда других групп элементов, в частности – и для переходных металлов. Это метод SCF.
Метод ZINDO/1 является вариантом метода INDO, адаптированного для проведения расчетов молекул, включающих атомы переходных элементов. Эквивалентен последней версии метода INDO/1, который отличается от оригинала использованием постоянных орбитальных экспонент. ZINDO/1 позволяет вычислять энергетику и геометрию молекул, содержащих переходные металлы.
Метод ZINDO/S является версией метода INDO, параметризованного для воспроизведения УФ и видимых оптических переходов при расчетах конфигурационного взаимодействия (CI) с одночастичными возбуждениями. Полезен для прогнозирования УФ и видимых спектров, но не пригоден для оптимизации геометрии или молекулярной динамики.
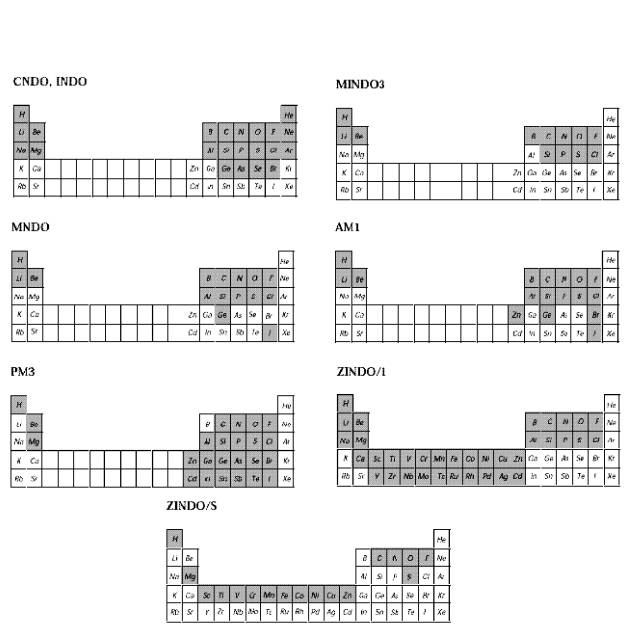
Для установки параметров полуэмпирических расчетов необходимо открыть диалоговое окно выбранного полуэмпирического метода. Есть две версии этого диалогового меню (смотри последующие разделы).
В этих таблицах представлены химические элементы, которые могут рассчитываться теми или иными полуэмпирическими методами. В том случае, если символ элемента присутствует в таблице, это означает, что в программе HyperChem для данного полуэмпирического метода существует часть для расчета интегралов с участием валентных электронов этого элемента с соответствующими главным и орбитальным квантовыми числами. Однако, в настоящее время не для всех атомов, для которых существует программная реализация того или иного полуэмпирического метода, известен набор параметров. Если таковые становятся известными, то можно изменять соответствующие файлы параметров. Элементы, для которых параметры известны и внесены в соответствующие файлы параметров, в этих таблицах закрашены.
Диалоговое
окно полуэмпирического метода
Диалоговое меню метода РМХ
В расширенном методе Хюккеля используется отличное от всех остальных полуэмпирических методов диалоговое окно.
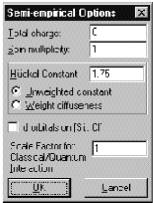
Total charge (Полный заряд системы) вычисляется как разность между полным количеством электронов в системе и суммарным зарядом ядер. Целочисленный и целочисленный положительный для катионов и отрицательный - для анионов.
Spin multiplicity (Мультиплетность по спину) вычисляется как 2S+1, где S – полный спин системы. Каждый неспаренный электрон имеет спин, равный 1/2. Системы с закрытой оболочкой (синглет) имеют мультиплетность, равную 1. Обладающие одним неспаренным электроном (дублет) и двумя (триплет) – 2 и 3 соответственно. В это диалоговое окно можно вводить величины от 1 до 6.
Huckel constant (Константа Хюккеля) пропорциональности между диагональными и недиагональными матричными элементами. Стандартное значение равно 1.75. Более высокие значения увеличивают вес перекрывания атомных орбиталей в определении полной энергии, а меньшие – одноэлектронных энергий.
Ö Unweightet constant (Не взвешенная константа) выбор этого пункта означает, что хюккелевская константа используется в расчетах без изменений.
Ö Weight diffuseness (Вес диффузности) умножает хюккелевскую константу на множитель, который учитывает диффузность атомных орбиталей, что встречается достаточно редко для органических молекул и молекул, состоящих и атомов главных подгрупп.
Ö d-orbitals on …(d-орбитали) этот пункт позволяет учитывать d-орбитали для атомов Si, P, S, Cl.
Scale factor (Масштабный
множитель) масштабирует введение классических частичных зарядов в
случае проведения смешанных (молекулярно-механических и квантово-химических)
расчетов (Для более детального ознакомления см. HyperChem Computation Chemistry, Theory and Methods).
Диалоговое окно других полуэмпирических методов
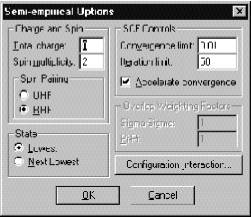
При выборе одного из полуэмпирических методов, перечисленных выше (исключая РМХ), возникает соответствующее диалоговое окно.
Charge and Spin
Ö Total charge (Полный заряд системы) вычисляется как разность между полным количеством электронов в системе и суммарным зарядом ядер. Целочисленный и целочисленный положительный для катионов и отрицательный - для анионов.
Ö Spin multiplicity (Мультиплетность по спину) вычисляется как 2S+1, где S – полный спин системы. Каждый неспаренный электрон имеет спин, равный ½. Системы с закрытой оболочкой (синглет) имеют мультиплетность, равную 1. Обладающие одним неспаренным электроном (дублет) и двумя (триплет) – 2 и 3 соответственно. В это диалоговое окно можно вводить величины от 1 до 6.
State (Состояние). Этот параметр описывает возбужденные состояния валентных электронов в системе.
Ö Lowest (Наинизшее) – выбор этого параметра означает, что программа будет выбирать низшее из всех возможных электронных состояний в системе с заданной мультиплетностью по спину.
Ö Next Lowest (Первое возбужденное) – выбор этого параметра означает, что программа будет рассчитывать первое возбужденное электронное состояние с заданной мультиплетностью по спину.
Ö Convergence Limit (Параметр сходимости). SCF расчет заканчивается тогда, когда отличия в полной энергии двух последующих итераций становятся меньше некоего заранее заданного значения. По умолчанию этому значению присваивается значение 0.01 ккал/моль. Этот параметр может меняться от 1 до 0.001. Параметр сходимости 1 ккал/моль является очень грубым, а 0.001 ккал/моль – не всегда достижим, так как систематическая ошибка полуэмпирических методов достигает примерно такой же величины. При поиске переходного состояния рекомендуется задавать минимальный параметр сходимости.
Ö Iteration limit (Предельное количество итераций). Этот параметр определяет предельное количество итераций на шаге самосогласования. Рекомендуемое количество – 50, но можно, в случае медленной сходимости, ставить и большее число – порядка 100 или 200, например – в случае поиска переходного состояния.
Ö Accelerate convergence (Ускорение сходимости). Выбор этого параметра убыстряет сходимость SCF расчетов. При этом HyperChem включает процедуру, известную как “Прямое инвертирование подпространства итераций” (Direct Inversion of Iterative Subspase, DIIS) (Более подробно смотри HyperChem Computational Chemistry).
Spin pairing (Спиновое состояние). Возможно выбрать два метода расчета спиновых состояний молекул. Первый – неограниченный метод Хартри-Фока (Unrestricted Hartree-Fock method, UHF) и ограниченный метод Хартри-Фока (Restricted Hartree-Fock method, RHF).
Ö UHF рассматривает спин-орбитали с различным пространственным распределением для a и b орбиталей. Этот метод применяется при изучении систем, как с открытыми, так и с закрытыми электронными оболочками. Так, для последних он хорошо описывает реакции диссоциации. Однако, из-за удвоения количества орбиталей, время расчета этим методом увеличивается вдвое. У этого метода существуют и другие ограничения, связанные с его основами.
Ö В RHF считается, что электроны с различным спином занимают одинаковые, в смысле пространственного распределения, орбитали. При этом неспаренные электроны тоже могут занимать отдельные орбитали. Этот метод применяется как для открытых, так и для закрытых электронных оболочек.
Overlap Weighting Factors (Коэффициент
масштабирования перекрывания). Дополнительные параметры для двух ZINDO
методов, которые способны изменять вклады σ и π
связей. Более подробно этот параметр описан в
HyperChem
Computational Chemistry, Theory and Methods.
Ö Sigma-Sigma определяет s-s перекрывание атомных орбиталей. Обычно он равен 1.0 для ZINDO/1 и 1.67 для ZINDO/S.
Ö Pi-Pi определяет вес s-s перекрывания атомных орбиталей. Он равен 1.0 для ZINDO/1. Для ZINDO/S этот параметр равен 0.640 при расчетах комплексов переходных металлов и 0.585 при расчетах органических молекул.
Configuration Interaction (Конфигурационное взаимодействие). Эта опция используется для активации расчета конфигурационных взаимодействий и открывает соответствующее диалоговое окно. Такой подход необходимо применять при расчетах УФ и оптических спектров в видимом диапазоне. Выбор этой опции существенно увеличивает время расчетов.
Диалоговое окно Configuration
Interaction
Учет конфигурационного взаимодействия может быть использован для улучшения качества волновой функции и энергии состояния. Все расчеты в приближении самосогласованного поля (SCF) основаны на одноэлектронной модели, суть которой заключается в том, что каждый электрон движется в усредненном поле, которое формируется всеми остальными электронами. Считается, что электроны взаимодействуют мгновенно и стремятся избегать друг друга согласно принципа Паули. Такая корреляция приводит к понижению среднего межэлектронного отталкивания и, в свою очередь, к понижению энергии состояния. Отличие между полной энергией, рассчитанной в SCF подходе и энергией, полученной в точно нерелятивистском подходе, называется корреляционной энергией.
Существуют два типа электронных корреляций: статические и динамические. Статические корреляции связаны с энергетическим вырождением данного состояния, а динамические – со стремлением электронов избегать друг друга, что происходит с бесконечно большой скоростью.
КВ (CI) расчеты, возможно, являются наиболее широко распространенным методом выхода за пределы SCF-подхода. Результатом SCF расчета является конфигурация состояния, в котором одноэлектронные уровни жестко заполнены электронами. Другие конфигурации могут быть сформированы из конфигурации, полученной в самосогласованном расчете при помощи возбуждения электронов с занятых на виртуальные (вакантные) орбитали. Результатом КВ расчета является набор улучшенных состояний, каждое из которых представляется линейной комбинацией таких конфигураций. КВ расчеты невозможно проводить в режиме оптимизации геометрии. В методе РМХ этот подход также не реализован.
Для установки параметров КВ расчетов используется диалоговое окно Configuration Interaction (Конфигурационного взаимодействия). Для этого необходимо выбрать соответствующую кнопку в диалоговом меню полуэмпирических методов.
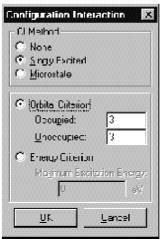
Затем нужно выбрать один из параметров: None (Ни одного), Singly Exited (Однократно возбужденное) или Microstate (Микросостояние).
Ö None - расчет конфигурационных взаимодействий производиться не будет.
Ö Singly Exited - в расчете будут учитываться только однократно возбужденные состояния.
Ö Microstate означает, что в расчете кроме однократно-возбужденных состояний будут учитываться и все возможные многократные.
Orbital Criterion (Орбитальный критерий). Выбор этого параметра определяет диапазон орбиталей, с которых и на которые происходят электронные возбуждения, формирующие взаимодействующие конфигурации.
Ö Occupied (Занятые) определяет область занятых орбиталей, начиная с высшей занятой молекулярной орбитали (HOMO), с которой происходит возбуждение орбиталей.
Ö Unoccupied (Вакантные) определяет область вакантных (виртуальных) орбиталей, начиная с низшей вакантной орбитали (LUMO), на которые происходят электронные возбуждения.
Energy Criterion (Энергетический критерий) является опцией Орбитального критерия, который устанавливает ограничения по энергии при генерировании набора взаимодействующих конфигураций. Эта опция доступна только для Однократно возбужденных конфигураций.
Ö Maximum Exitation (Максимальное возбуждение) определяет наибольшую разницу по энергии в эВ между занятыми и вакантными орбиталями, включенными в CI расчет. В общем виде, конфигурации с высокой энергией не могут сильно взаимодействовать с конфигурацией основного состояния. Чем выше этот параметр, тем больше конфигураций включается в CI расчет.
Практические применения метода конфигурационного взаимодействия.
CI расчеты возможно использовать при расчетах:
q УФ и видимых спектров,
q Энергии возбужденных состояний,
q Изучения создания или разрыва химических связей (например, диссоциация H2), изменения спинового состояния,
q Изучения эффектов, связанных с дисперсионными силами Лондона,
q Описания вырожденных, или близких к вырождению состояний,
q Изучения расщепления синглет-триплет на более высоком уровне.
Метод микросостояний понижает энергию некоррелированного состояния так же, как и возбужденных состояний. Метод однократно возбужденного CI предназначен только для расчетов УФ и видимых спектров и не улучшает энергию. Основного состояния (теорема Бриллюэна).
При использовании орбитального критерия для симметричных систем, для того, чтобы получить корректные результаты, необходимо включить либо все, либо ни одного из наборов вырожденных орбиталей. Необходимо также внимательно использовать Энергетический критерий. Этот критерий должен быть больше, нежели чем энергетическая щель между занятыми и вакантными орбиталями.
В больших системах, как правило, в небольших энергетических интервалах находится большое количество орбиталей. Следовательно, размер CI матрицы может быть очень чувствительным к величине энергетического критерия. Так как время вычислений сильно зависит от размера CI матрицы, необходимый вычислительный ресурс, особенно если использовать методы MNDO, AM1 или PM3, может стать не приемлемо большим. Для того чтобы избежать такой ситуации, необходимо тщательно проанализировать результаты RHF расчета.
Для того чтобы более детально ознакомится со спецификой расчета УФ и оптических спектров, необходимо ознакомится с “Electronic Spectrum” на стр. 285 файла Referenc.pdf
![]()
![]()
![]()