![]()
![]()
![]()
Глава II.6.4.
Неэмпирический
(ab initio) метод Хартри-Фока
Выбор параметра ab initio в меню Setup позволяет проводить неэмпирические расчеты электронной и атомной структур объектов. В отличие от молекулярно-механических и полуэмпирических методов, неэмпирический метод Хартри-Фока не требует для проведения расчетов знания каких-либо эмпирических параметров, например – силы и длинны отдельных связей, значений интегралов перекрывания и пр. В меню Setup этот пункт стоит третьим.
Ab initio метод требует для своих расчетов гораздо больше вычислительных ресурсов, нежели чем молекулярно-механические и полуэмпирические методы. Особенно это касается оптимизации геометрии или проведения молекулярно-динамических расчетов. Для оптимизации геометрии рекомендуется, на начальном этапе использовать молекулярную механику, затем – один из полуэмпирических методов, для того, чтобы получить более или менее обоснованную начальную геометрию. Однако для ряда неорганических систем молекулярно-механические и полуэмпирические расчеты дают некорректные результаты, поэтому рекомендуется использовать параметр Model Bilder, для того, чтобы получить более или менее подходящую стартовую геометрию.
Выбор
базисного набора
Любой набор одноэлектронных волновых функций может служить базисным набором (или просто – базисом) для ЛКАО (линейная комбинация атомных орбиталей, английская аббревиатура – LCAO) приближения. Однако, хорошо определенный базис будет предсказывать электронные свойства системы с использованием гораздо большего числа членов, нежели, чем плохо определенный. Следовательно, выбор наиболее подходящего базисного набора в ab initio расчете является критичным для точности и обоснованности результатов. В программе HyperChem определен формат файла базисных наборов (расширение *.BAS), в который включены целый ряд стандартных базисных наборов. Тем не менее, пользователь может сам определить необходимые для расчетов базисные наборы.
Много обычных и широко используемых базисных наборов автоматически поддерживаются в HyperChem. Эти наборы включают в себя:
• STO-1G and STO-1G* (H до He) [1] ;
• STO-2G and STO-2G* (H до Xe) [1] ;
• STO-3G and STO-3G* (H до Xe) [1] ;
• STO-4G and STO-4G* (H до Xe) [1] ;
• STO-5G and STO-5G* (H до Xe) [1] ;
• STO-6G and STO-6G* (H до Xe) [1] ;
• 3-21G, 3-21G*, and 3-21G** (H до Ar) [2] ;
• 4-21G, 4-21G*, and 4-21G** (H до Ne) [3] ;
• 6-21G, 6-21G*, and 6-21G** (H до Ar) [2] ;
• 4-31G, 4-31G*, and 4-31G** (H до Ne) [3] ;
• 5-31G, 5-31G*, and 5-31G** (H до F) [3] ;
• 6-31G, 6-31G*, and 6-31G** (H до Ar) [3] ;
• 6-311G, 6-311G*, and 6-311G** (H до Ar) [4] ;
• D95, D95* and D95** (H до Cl) [5] .
Диалоговое
окно метода ab initio
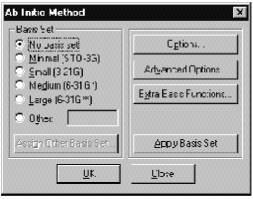
В программе HyperChem возможно использовать много базисных наборов. В этом диалоговом окне кнопка Apply Basis Set служит для того, чтобы установить выбранный базис или для всего объекта, или для выделенной части, если такое выделение было сделано. Например, некоторые тяжелые атомы должны описываться базисом 6-31G (без d-функций), тогда как другие – базисом 6-31G* (с учетом d-функций). Параметр Basis Set диалогового меню приписывает соответствующий базис или всей молекуле, либо выделенной части.
Ö Выбор параметра NoBasis Set означает, что данному атому не будет приписываться ни одной базисной функции. Эта опция может быть использована только в том случае, если необходимо описать систему или выделенную часть с использованием дополнительных базисных функций.
Ö Minimal (STO-3G) приписывает минимальный STO-3G базис. Другие кнопки этого меню выбирают те базисные наборы, которые там указаны.
Ö Other позволяет активизировать кнопку Assign Other Basis Set, для того, чтобы использовать другие (не обозначенные в этом меню) базисные наборы.
Assign Other Basis Set. Нажатие этой кнопки приводит к вызову соответствующего меню, которое содержит полный список базисных наборов (исключая те, которые были приведены в предыдущем меню).
Extra Basis Function (Дополнительные базисные функции). Нажатие этой кнопки приводит к появлению соответствующего меню, которое позволяет вводить дополнительные базисные функции для выбранных атомов.
Options (Параметры расчета). Нажатие этой кнопки приводит к вызову соответствующего диалогового окна, при помощи которого задаются основные параметры ab initio расчета.
Advanced options служит для вызова соответствующего меню, в котором содержатся параметры, влияющие на процесс расчетов.
Кнопка Aply Basis Set присваивает всей молекуле или выделенной части атомов выбранный ранее базис. Кнопка OK служит для сохранения выбранных параметров и закрытия диалогового окна. Кнопка Cancel приводит к закрытию диалогового окна без сохранения выбранных параметров. Как уже говорилось ранее, в программе HyperChem существует возможность определять различные базисные наборы для разных частей рассчитываемой системы.
Диалоговое
окно выбора других базисных наборов (Assign Other Basis Set Dialog Box)

Использование этого диалогового окна позволяет вызывать полный список базисных наборов, исключая те, которые уже были обозначены в диалоговом окне Ab Initio Method.
Выбор любого из этих базисов и последующее нажатие на кнопку OK приведет к тому, что в соответствующем окне Ab Initio Method появится текстовая идентификация сделанного выбора. Все стандартные базисные наборы, как это уже говорилось ранее, хранятся в соответствующих файлах с расширением .BAS. Для того чтобы создать соответствующую ссылку на свой базисный набор в этом диалоговом окне, необходимо создать для него точку входа в секции [basisset] Registry или в соответствующем файле CHEM.INI.
Диалоговое
окно параметров ab initio (Options)
Это диалоговое окно используется для выбора основных параметров неэмпирических вычислений. Эти параметры аналогичны параметрам полуэмпирических методов, описание которых было дано выше.
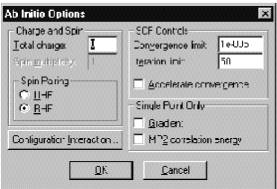
Ö Gradient задает расчет градиентов (первых производных полной энергии по атомным координатам). RMS градиент дает представление об отклонениях от оптимальной геометрии рассчитываемого объекта. Эта опция доступна только в режиме расчета одной точки. Это связано с тем, что для расчета этих параметров необходимо рассчитывать много двухэлектронных интегралов и их производных, что требует много процессорного времени, а в этом режиме необходимость таких расчетов может отсутствовать. Процедура HyperGauss, которая осуществляет неэмпирические расчеты, всегда рассчитывает градиенты при оптимизации геометрии, при молекулярно-динамических расчетах и расчетах молекулярных колебаний.
Ö MP2 Correlation Energy задает расчет корреляционной энергии в рамках теории возмущения Меллера-Плессета второго порядка. Эта опция тоже активна только для расчетов одной точки. Такие расчеты тоже увеличивают процессорное время, необходимую оперативную память и используемое дисковое пространство, так как для этого требуется переход от двухэлектронных интегралов на атомных орбиталях к таковым, рассчитанным уже на молекулярных орбиталях.
Это диалоговое окно имеет также параметры Charge и Spin multiplicity. Они служат для того, чтобы задавать полный электронный заряд системы, который определяется как разность между количеством электронов и суммарным ядерным зарядом и мультиплетность системы, которая определяется как 2S+1 и может быть синглетом (1), дублетом (2), триплетом (3) и квартетом (4).
SCF Controls (Секция параметров SCF расчета)
Эта часть диалогового меню служит для задания требуемой точности расчета SCF волновой функции и максимального количества итераций для достижения этой точности.
Ö Convergence (Limit Предел сходимости) служит для того, чтобы остановить SCF процедуру, когда разница в энергиях между двумя последующими итерациями становится меньше заданной величины. Для ab initio расчетов это значение, как правило, выбирается равным 0.00001 ккал/моль, а используемый параметр может лежать в интервале от 1 до 0.00000001 ккал/моль. Тем не менее, если этот параметр задать меньше 10**-10, то сходимости можно не достигнуть, так как сам метод Хартри-Фока дает соразмерную ошибку. При поиске переходных состояний рекомендуется использовать более жесткие критерии сходимости.
Ö Iteration Limit (Предельное количество итераций) определяет максимальное количество итераций в SCF расчете. Расчет останавливается, в случае если программа выполнила заданное количество итераций и при этом не достигла сходимости. В этом случае результат расчета может быть не верен, так как энергия может быть далека от истинной, либо она осциллировала в ходе расчета. 50 итераций представляется разумным для большинства случаев, тогда как задание большего количества (скажем, до 200) может быть оправданным при расчетах переходных состояний. В случае, когда расчет не может сойтись и при большем количестве итераций, то это, как правило, означает, что дальнейшее увеличение количества итераций не может изменить, так как система не сходится. Лишь в некоторых случаях Ö Accelerate Convergence (Алгоритм ускорения сходимости) может исправить ситуацию.
Выбор этого параметра убыстряет сходимость SCF расчетов. При этом HyperChem включает процедуру, известную как “Прямое инвертирование подпространства итераций” (Direct Inversion of Iterative Subspase, DIIS) (Более подробно смотри HyperChem Computational Chemistry), которая может потребовать большего объема памяти. Включение этой опции может увеличить время, затрачиваемое на одну итерацию из-за того, что в этом подходе матрица Фока вычисляется как линейная комбинация текущей матрицы Фока и матриц Фока от предыдущих итераций. Как правило, такой подход уменьшает общее количество требуемых итераций.
Spin pairing (Спиновое состояние). Возможно выбрать два метода расчета спиновых состояний молекул. Первый – неограниченный метод Хартри-Фока (Unrestricted Hartree-Fock method, UHF) и ограниченный метод Хартри-Фока (Restricted Hartree-Fock method, RHF).
Ö UHF рассматривает спин-орбитали с различным пространственным распределением для a - и b- орбиталей. Этот метод применяется при изучении систем, как с открытыми, так и с закрытыми электронными оболочками. Так, для последних он хорошо описывает реакции диссоциации. Однако, из-за удвоения количества орбиталей, время расчета этим методом увеличивается вдвое. У этого метода существуют и другие ограничения, связанные с его основами.
Ö В RHF считается, что электроны с различным спином занимают одинаковые, в смысле пространственного распределения, орбитали. При этом неспаренные электроны тоже могут занимать отдельные орбитали. Этот метод применяется как для открытых, так и для закрытых электронных оболочек.
Configuration Interaction (Конфигурационное взаимодействие). Эта опция используется для активации расчета конфигурационных взаимодействий и открывает соответствующее диалоговое окно. Такой подход необходимо применять при расчетах УФ и оптических спектров в видимом диапазоне. Выбор этой опции существенно увеличивает время расчетов.
Кнопка OK служит для сохранения выбранных параметров и закрытия диалогового окна. Кнопка Cancel приводит к закрытию диалогового окна без сохранения выбранных параметров. Как уже говорилось ранее, в программе HyperChem существует возможность определять различные базисные наборы для разных частей рассчитываемой системы.
Необходимо отметить, что HyperChem не поддерживает ограниченный метод Хартри-Фока для систем с открытыми оболочками (ROHF) на ab initio уровне.
Диалоговое
окно параметров высокого уровня метода ab initio (Ab Initio Advanced options)
Это диалоговое окно служит для более точной настройки параметров ab initio расчетов.
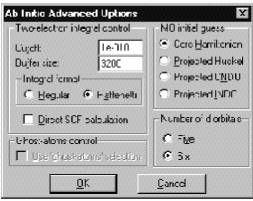
Integral Format
Ö Regular определяет использование обычного формата для записи двухэлектронных интегралов. HyperChem использует 16 байт для записи каждого из интегралов. Первые 8 байт хранят 4 индекса интеграла, а последние 4 – его значение. Программа сохраняет эти величины только в том случае, если абсолютное значение интеграла больше или равно параметру Cutoff. В противоположном случае значение этого интеграла приравнивается к 0. Двухэлектронные интегралы и их индексы сохраняются на диске без модификации при выборе опции Regular и могут быть записаны в .log-файл при соответствующем выборе параметра QuantumPrintLevel меню StartLog.
ÖRaffenetti определяет использование формата Раффенети (R.C. Raffenetti, Chem.Phys.Lett., 20, 335 (1973)), который позволяет более просто формировать матрицу Фока в ходе SCF расчета. Этот формат, как правило, требует больше памяти и больше дискового пространства, однако позволяет повышать скорость расчета. Этот формат нельзя использовать при проведении MP2-расчетов.
Параметр Cutoff позволяет сохранять на диске только те интегралы, абсолютное значение которых равно или превышает задаваемый параметр. По умолчанию он равен 10**-10 Хартри. Этот параметр контролирует осуществление SCF-итераций, точность волновых функций и энергии, так как он уменьшает количество рассчитываемых двухэлектронных интегралов.
Параметр Buffer size (Размер буфера) определяет размер операционной памяти (в словах двойной точности, 8 байт для одного слова), которая требуется для хранения двухэлектронных интегралов до того, как записать их во временный на жестком диске (выбор этого диска может быть сделан в меню File/Preferences/Path). Более большой размер буфера способен уменьшить расчетное время из-за того, что программа будет обращаться к диску более редко. Если этот буфер будет достаточно велик, то HyperChem не будет обращаться к диску вовсе. Необходимо отметить, что в случае, когда программа останавливается из-за сбоя компьютера, либо по другой некорректной причине, этот временный файл остается на диске. Для того чтобы освободить это место, его необходимо удалять вручную.
Direct SCF calculation вычисляет двухэлектронные интегралы на каждой итерации, а не один раз перед SCF шагом, как если бы это было без нее. Такой расчет, безусловно, гораздо медленнее, но он позволяет не использовать дисковое пространство и операционную память под большое количество интегралов. Включение этой опции требуется при расчетах больших систем на компьютерах с маленьким диском и памятью.
Ghost-atoms Control (Использование атомов-призраков). Эта опция позволяет вводить центры, которым приписываются базисные функции тех или иных атомов, при этом в систему не вводятся ни дополнительные ядра, ни дополнительные электроны. Эта опция позволяет вводить только дополнительные базисные функции, центрированные в любой точке пространства. Необходимо помнить, что использование таких “виртуальных” атомов может приводить к неким артефактам. Например, так как существуют базисные волновые функции, будут существовать и мулликеновские заряды, которые будет центрированы на соответствующих центрах. Для того чтобы использовать эту возможность, необходимо в меню Select, предварительно сделав соответствующее выделение, выбрать пункт Name selection и, выбрав пункт Other, приписать ему имя ghost-atoms. После этого в диалоговом меню Ab Initio Method / Advanced Options становится активным пункт Ghost-atoms Control. Такой подход бывает эффективным при описании ряда неординарных химических связей. Эта опция активна только для расчетов одной точки.
MO initial guess (Параметр стартового заселения МО) определяет стартовое заполнение коэффициентов молекулярных орбиталей при помощи диагонализации остового гамильтониана. При выборе параметра Projected Huckel, эти параметры определяются по методу Хюккеля. Аналогично определяются и первоначальные коэффициенты при выборе параметров Projected CNDO (методом CNDO) и Projected INDO (методом INDO).
Number of d Orbitals (Количество d-орбиталей). Этот параметр определяет вид d-орбиталей, используемых в расчете. Выбор пяти (five) орбиталей соответствует расчету с использованием эрмитовых орбиталей (d 0, d 1, d –1, d 2, d –2), а выбор шести (six) – соответствует расчету с использованием d-орбиталей в декартовом представлении (d xx, d yy, d zz, d xy, d xz, d yz).
Кнопка OK служит для сохранения выбранных параметров и закрытия диалогового окна. Кнопка Cancel приводит к закрытию диалогового окна без сохранения выбранных параметров.
С другими особенностями программы HyperChem читатель может познакомиться, изучив файлы Referenc.pdf, CDK.pdf и GetStart.pdf.
Меню Compute определяет вид расчета. Это может быть расчет в одной точке (Single Point), оптимизация геометрии (Geometry Optimization), молекулярная динамика (Molecular Dynamics), ланжевеновская динамика (Langevin Dynamics), расчеты методом Моне-Карло, а также некоторые другие виды расчетов. Далее по тексту книги нас в основном будут интересовать результаты расчетов методом молекулярной динамики, которые представлены файлами с динамическим кино. Для того чтобы просмотреть результаты молекулярно-динамического моделирования, необходимо прочитать соответствующий файл (в тексте они будут указываться), а затем, выбрав в меню Compute пункт Molecular Dynamics, отметить Ö Playback и нажать Proceed. Для увеличения скорости проигрывания файлов (когда это необходимо, в зависимости от сложности задачи и мощности компьютера) можно увеличить time steps (по умолчанию стоит 1, при увеличении скорость увеличивается кратно количеству шагов).
Как уже говорилось выше, подробная инструкция о пользовании программой HyperChem дается в файлах CDK.pdf, GetStart.pdf, Referenc.pdf. Но научиться с ней работать не составляет большого труда, так как программа сконструирована очень удобно и предназначена для широкого круга пользователей с разным уровнем квалификации.
Для желающих более детально ознакомится с некоторыми приемами и алгоритмами компьютерной химии, авторы могут порекомендовать хороший и легко доступный по пониманию источник [6], в котором они приведены.
![]()
![]()
![]()