1.3.2. Многочастичные потенциалы
1.3.2.1. Потенциал
Стиллингера


Потенциал Стиллингера
Вебера
(Stillinger
Weber)
[66] является одним из первых потенциалов, описывающих
материалы, образующие алмазоподобную структуру (C, Si, GaAs, Ge). Потенциальная
энергия этого потенциала записывается в следующем виде:
|
Здесь θijk
угол,
центрированный на атоме i, RC
радиус
обрезания потенциала.
При β = cos(109,47°) =
1/3
энергетически выгодной является алмазоподобная конфигурация. Потенциал
Стиллингера
Вебера
является достаточно популярным и часто применяется в численных расчетах кристаллического
кремния [67, 68].
Основной проблемой данного потенциала является его плохая
переносимость, поскольку трехчастичный вклад в (1.174) имеет только одну равновесную конфигурацию при β =
1/3.
Однако в случае углерода, кроме 109,47°, стабильными являются
конфигурации с углами 180° и 120°. Также данный потенциал дает неверные результаты в
случае его использования для оптимизации поверхности кристалла кремния и кремниевых
кластеров малых размеров.
Этих недостатков лишен потенциал Абеля
Терсоффа
Бреннера,
разработанный для углерода и его соединений с водородом и основанный на
зависимости прочности связи от локального атомного окружения.
Таблица 1-5.
Параметры потенциала Стиллингера
Вебера
для кремния
A |
B |
p |
q |
RC |
λ |
γ |
7,049556277 |
0,6022245584 |
4 |
0 |
1,8 |
21 |
1,2 |
1.3.2.2. Потенциал
Абеля
Общая форма этого потенциала была предложена Абелем (Abel) [69], после чего он был доработан Терсоффом (Tersoff) [70] и впоследствии параметризован для углерода и его
соединений с водородом Бреннером (Brenner) [71, 72]. Данный потенциал широко используется для
молекулярно-динамических расчетов углеродных соединений (таких как углеродные
кластеры, нанотрубки и т. п.) [73
78]. Потенциал Абеля
Терсоффа
Бреннера
(ПАТБ) хорошо описывает механические свойства подобных структур, упругие свойства
и зависимость динамики данных структур от температуры.
Потенциальная энергия, полученная с использованием ПАТБ, записывается в следующем виде:
|
где rij
расстояние
между ближайшими соседними атомами i и j, bij
порядок
связи между атомами i и j:
|
Значение функции
зависит от атомного окружения и угла связи для
атома i. Функция
также состоит из двух членов:
|
Выражения (1.176) и (1.177), скомбинированные с (1.175), используются для определения энергии связи из-за ковалентного связывания любого набора атомов углерода и водорода.
Член, связанный с межатомным отталкиванием и притяжением (1.175), записывается в следующем виде:
|
|
|
(1.179) |
Экранированная кулоновская функция
используется для описания парного отталкивания в (1.178), а функция
ограничивает радиус ковалентного
взаимодействия. В случае описания углеродной системы предполагается, что
функция
равна единице для ближайших соседей и нулю в
любом другом случае.
Первый член в уравнении (1.176) записывается в следующем виде:
|
где функция P
это
бикубический полином, а числа
и
это
количество соседних атомов углерода и кислорода. Они определяются следующим
образом:
|
(1.181) |
|
(1.182) |
Функция
в (1.180) определяет вклад в порядок связи между атомами i и j со стороны соседнего атома k как косинус угла j
i
k (табл. 1-6, 1-7,
рис. 1-12, 1-13). Аналитический вид функции
для углерода был получен как полином шестого
порядка. Вся информация для конструирования этого полинома дана в
табл. 1-6.
Таблица 1-6. Параметры для углового вклада
|
||||
θ, рад |
|
|
|
γ(θ) |
0 |
8 |
|
1 |
|
π/3 |
2,0014 |
|
0,416335 |
|
π/2 |
0,37545 |
|
0,271856 |
|
0,6082π |
0,09733 |
0,40000 |
1,98000 |
|
2π/3 |
0,05280 |
0,17000 |
0,37000 |
|
π |
|
0,10400 |
0,00000 |
|
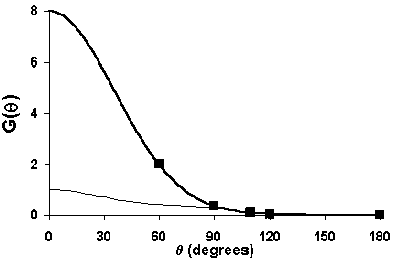
Рис. 1-12. Вид полиномов GC(θ) (жирная
линия) и γC(θ) (тонкая линия). Отмеченные точки
соответствуют значениям, приведенным в табл. 1-6.
Таблица 1-7.
Параметры для углового вклада
|
||||||
θ (рад) |
0 |
π/3 |
π/2 |
2π/3 |
5π/6 |
π |
|
19,991787 |
19,704059 |
19,065124 |
16,811574 |
12,164186 |
11,235870 |
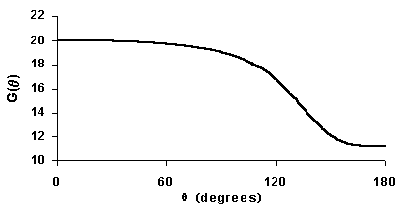
Рис. 1-13. Вид полинома GH(θ).
Полином γ(θ) должен быть использован для систем с избыточным и недостаточным числами соседей. Таким образом, общая угловая функция для углов, больших 0, но меньших, чем 109,47°, для атома углерода i имеет следующий вид:
|
Общая угловая функция для атома водорода имеет вид
.
Функция Q в (1.183) определяется как
|
(1.184) |
Здесь
координационное
число атома i:
|
(1.185) |
Первый член в (1.177) зависит от полного числа соседей связанных атомов i и j и от функции
,
определяющей, является ли связь частью сопряженной системы:
|
где
|
(1.187) |
и
|
Если все атомы углерода связаны в пары, i и j имеют четыре соседа или более, то уравнения (1.186)
(1.188) определяют
равным единице. Таким образом, связь между
этими атомами не является частью сопряженной системы. При уменьшении координационных
чисел соседствующих атомов
становится больше единицы, показывая, что
связь становится частью сопряженной системы. Например, значение Nconj для
углерод-углеродной связи в графите равно девяти, в то время как для бензола Nconj = 3.
Значения F даны в табл. 1-8. Интерполяцию между ними следует
проводить с помощью трикубического полинома (табл. 1-9).
Таблица 1-8. Узловые значения функции FCC. Все значения и производные, не приведенные в таблице, считаются равными нулю. Все производные вычисляются как симметричные численные разности. F(i, j, k) = F(j, i, k), F(i, j, k > 9) = F(i, j, 9), F(i > 3, j, k) = F(3, j, k), F(I, j > 3, k) = F(i, 3, k) |
|||||||
i |
j |
k |
FСС(i, j, k) |
i |
j |
k |
FСС(i, j, k) |
1 |
1 |
1 |
0,10500000 |
0 |
1 |
2 |
0,0099172158 |
1 |
1 |
2 |
|
0 |
2 |
1 |
0,0493976637 |
1 |
1 |
3-9 |
|
0 |
2 |
2 |
|
2 |
2 |
1 |
0,09444957 |
0 |
3 |
1
|
|
2 |
2 |
2 |
0,02200000 |
1 |
2 |
1 |
0,0096495698 |
2 |
2 |
3 |
0,03970587 |
1 |
2 |
2 |
0,0300000000 |
2 |
2 |
4 |
0,03308822 |
1 |
2 |
3 |
|
2 |
2 |
5 |
0,02647058 |
1 |
2 |
4 |
|
2 |
2 |
6 |
0,01985293 |
1 |
2 |
5 |
|
2 |
2 |
7 |
0,01323529 |
1 |
2 |
6
|
|
2 |
2 |
8 |
0,00661764 |
1 |
3 |
2
|
|
2 |
2 |
9 |
0,00000000 |
2 |
3 |
1
|
|
0 |
1 |
1 |
0,04338699 |
||||
Производная |
Значение |
Производная |
Значение |
|
|
|
0,062418 |
|
|
|
|
|
0,000000 |
|
|
|
0,062418 |
|
|
|
|
Таблица 1-9. Узловые значения трикубического полинома FCH. Все значения и производные, не приведенные в таблице, считаются равными нулю. F(i, j, k) = F(j, i, k), F(i, j, k > 9) = F(i, j, 9) |
|||||||
i |
j |
k |
FCH(i, j, k) |
i |
j |
k |
FCH(i, j, k) |
0 |
2 |
5
|
−0,0090477875161288110 |
1 |
2 |
1
|
|
1 |
3 |
1
|
|
1 |
1 |
1
|
|
Последний член в (1.177) имеет следующий вид:
|
где
|
(1.190) |
Здесь функция
трикубический
полином (табл. 1-9); ejik и eijl
единичные
векторы, направленные параллельно векторным произведениям
и
соответственно, где
вектор,
соединяющий атомы i и j.
Значения функции TC-C даны в табл. 1-10, параметры для уравнений (1.178) и
(1.191)
в
табл. 1-11
1-14 .
Таблица 1-10. Узловые значения для углерод-углеродного кубического полинома TСС в уравнении (1.189). Все значения, не приведенные в таблице, считаются равными нулю. T(i > 3, j, k) = T(3, j, k), T(i, j > 3, k) = T(i, 3, k), T(i, j, k > 9) = T(i, j, 9) |
|||
i |
j |
k |
TСС(i, j, k) |
2 |
2 |
1 |
|
2 |
2 |
9 |
|
Функция, определяющая радиус ковалентного взаимодействия, имеет следующий вид:
|
где разность
определяет расстояние, на котором функция
меняется от единицы да нуля.
Таблица 1-11. Параметры для углерод-углеродных членов в уравнениях (1.178) и (1.191) |
||
B1 = 12388,79197798 эВ |
β1 = 4,7204523127 Å
|
Q = 0,3134602960833 Å |
B2 = 17,56740646509 эВ |
β2 = 1,4332132499 Å
|
A = 10953,544162170 эВ |
B3 = 30,71493208065 эВ |
β3 = 1,3826912506 Å
|
α = 4,7465390606595 Å-1 |
Dmin = 1,7 Å |
Dmax = 2,0 Å |
|
Таблица 1-12. Параметры для водород-водородных членов в уравнениях (1.178) и (1.191). Все значения функций F, Bi и βi равны нулю, кроме приведенных в таблице |
||
B1 = 29,632593 эВ |
β1 = 1,71589217 Å
|
Q = 0,370471487045 Å |
λHHH = 4 |
FHH(1,1,1) = 0,249831916 |
A = 32,817355747 эВ |
Dmin = 1,1 Å |
Dmax = 1,7 Å |
α = 3,536298648 Å
|
Таблица 1-13. Параметры для углерод-водородных членов в уравнениях (1.178) и (1.191). Все значения функций Bi и βi равны нулю, кроме приведенных в таблице |
||
B1 = 32,3551866587 эВ |
β1 = 1,43445805925
Å
|
Q = 0,340775728 Å |
Dmin = 1,3 Å |
Dmax = 1,8 Å |
A = 149,94098723 эВ |
α = 4,10254983 Å
|
||
Таблица 1-14. Узловые значения для углерод-углеродного и углерод-водородного бикубического полинома P. Все значения, не приведенные в таблице, считаются равными нулю |
|||||
i |
j |
PCC(i, j) |
i |
j |
PCH(i, j) |
1 |
1 |
0,003026697473481 |
1 |
0 |
0,209336732825038 |
2 |
0 |
0,007860700254745 |
2 |
0 |
−0,064449615432525 |
3 |
0 |
0,016125364564267 |
3 |
0 |
−0,303927546346162 |
1 |
2 |
0,003179530830731 |
0 |
1 |
0,010000000000000 |
2 |
1 |
0,006326248241119 |
0 |
2 |
−0,1220421462782555 |
1 |
1 |
−0,1251234006287090 |
|||
2 |
1 |
−0,2989052457830000 |
|||
0 |
3 |
−0,3075847050660000 |
|||
1 |
2 |
−0,3005291724067579 |
|||
1.3.2.3. Потенциал
Клери
Потенциал Клери
Росато
(Cleri
Rosato) [79]
может быть применен для описания переходных металлов (ГЦК и ГПУ) и сплавов
(табл. 1-15, 1-16). Данный потенциал корректно описывает динамику решетки
этих материалов, а также их поведение при ненулевой температуре.
Энергия связи системы записывается в следующем виде:
|
где rij
расстояние
между текущим атомом i и атомом j;
расстояние
до ближайшего соседа; ξ
эффективный
интеграл перескока; q описывает зависимость ξ от относительного межатомного расстояния. ξ и q предполагаются
зависящими только от типов взаимодействующих атомов α и β.
Суммирование по j в уравнении (1.192) идет вплоть до соседей пятого порядка в кубической структуре и до соседей девятого порядка в гексагональной плотноупакованной структуре.
Таблица 1-15.
Параметры для потенциала Клери
|
||||
Металл |
A, эВ |
ξ, эВ |
p |
Q |
Ni |
0,0376 |
1,070 |
16,999 |
1,189 |
Cu |
0,0855 |
1,224 |
10,960 |
2,278 |
Rh |
0,0629 |
1,660 |
18,450 |
1,867 |
Pd |
0,1746 |
1,718 |
10,867 |
3,742 |
Ag |
0,1028 |
1,178 |
10,928 |
3,139 |
Ir |
0,1156 |
2,289 |
16,980 |
2,691 |
Pt |
0,2975 |
2,695 |
10,612 |
4,004 |
Au |
0,2061 |
1,790 |
10,229 |
4,036 |
Al |
0,1221 |
1,316 |
8,612 |
2,516 |
Pb |
0,0980 |
0,914 |
9,576 |
3,648 |
Для переходных металлов, образующих гексагональную
плотноупакованную структуру, необходимо ввести новый параметр
(где c и a
параметры
элементарной ячейки).
Таблица 1-16.
Параметры для потенциала Клери
|
|||||
Металл |
A, эВ |
ξ, эВ |
p |
q |
β |
Ti |
0,1519 |
1,8112 |
8,620 |
2,390 |
1,5874 |
0,0741 |
1,4163 |
11,418 |
1,643 |
1,6354 |
|
Zr |
0,1934 |
2,2792 |
8,250 |
2,249 |
1,5925 |
0,0523 |
1,4489 |
13,940 |
1,071 |
1,6409 |
|
Co |
0,0950 |
1,4880 |
11,604 |
2,286 |
1,6232 |
Cd |
0,1420 |
0,8117 |
10,612 |
5,206 |
1,8856 |
0,0416 |
0,4720 |
13,639 |
3,908 |
1,6511 |
|
Zn |
0,1477 |
0,8900 |
9,689 |
4,602 |
1,8562 |
Mg |
0,0290 |
0,4992 |
12,820 |
2,257 |
1,6235 |
Кроме вышеописанного, было предложено множество других эмпирических
потенциалов: для гранецентрированных металлов были разработаны потенциалы
«Сандиа» («Sandia»)
(Au, Cu, Pd, Ag, Pt, Ni, Al, некоторые сплавы) [80
82], Джонсона (Johnson) для большинства подобных металлов [83] и их сплавов [84], «стыковочные» («glue») (Au, Al) [84
87], потенциалы Вотера
Чена
(Voter
Chen) для Al и Ni [88], для сплава Ni3Al [89]. Для металлов с гексагональной плотноупакованной
структурой были разработаны потенциалы Оха
Джонсона
(Oh
Johnson) (Mg, Ti, Zr) [90], Пасионато
Савино
(Pasionat
Savino) (Hf, Ti, Mg, Co) [91]. Для ОЦК металлов были предложены следующие потенциалы:
потенциал Финниса
Синклера
(Finnis
Sinclair)
(Fe, V, Nb, Ta, Mo, W) [92, 93] и Оха
Джонсона
(Li, Na, K, V, Nb, Ta, Cr, Mo, W, Fe) [94].