1.3.3 Метод молекулярной динамики
Молекулярная динамика (МД)
это
техника компьютерного моделирования, позволяющая проследить эволюцию системы
взаимодействующих атомов во времени с помощью интегрирования уравнений
движения. Несомненным преимуществом метода МД является возможность
моделирования системы при заданной температуре или при заданных скоростях
атомов (ионов) с достаточно высокой скоростью расчета.
Сейчас МД применяется в квантовой химии и физике твердого тела для [95]:
· изучения дефектов в кристаллах, варьирующихся от точечных (вакансии, дефекты внедрения) до линейных (дислокации) и плоских (межфазные, междоменные границы и т. д.). Для расчета подобных структур нужно применять метод суперячеек, требующих использования большого количества атомов в системе [96];
· реконструкции поверхности кристалла, связанной с перестройкой координат множества атомов на поверхности. Ее можно описать с помощью МД, причем можно проследить изменение поверхности в зависимости от температуры [97];
· изучения кластеров, величина которых варьируется от нескольких атомов до нескольких тысяч, С помощью МД в основном проводится процедура численного отжига при оптимизации геометрии [98, 99];
· изучения биологических молекул (белки, ДНК, РНК и др.), обладающих низкой симметрией и содержащих огромное количество атомов [100].
1.3.3.1 Классическая молекулярная динамика
В классической молекулярной динамике используются законы механики Ньютона. Сила, действующая на атом I, записывается, соответственно, в виде
|
(1.193) |
где MI
масса
атома I,
его
ускорение. В расчетах классической МД используются эмпирические потенциалы (см.
выше).
Уравнения движения
Рассмотрим системы N частиц, двигающихся под влиянием потенциальной функции U [101]. Движения частиц можно описать с помощью их
координат
и импульсов
.
Обозначим
и
как
и
соответственно. Потенциальная функция U зависит только от координат
частиц:
.
Гамильтониан такой системы имеет следующий вид:
|
(1.194) |
Силу, действующую на частицу, можно вычислить как пространственную производную потенциала:
|
(1.195) |
Таким образом, уравнения движения такой системы можно записать в следующем виде:
|
(1.196) |
|
(1.197) |
Из этих уравнений легко получить второй закон Ньютона:
|
(1.198) |
Уравнения движения также можно получить, используя формализм Лагранжа:
|
(1.199) |
Последнее выражение применяется для получения ab initio МД-уравнений.
Интегрирования уравнений движения, алгоритм Верле
Для того чтобы проследить эволюцию системы, необходимо провести интегрирование уравнений движения, т. е. на основе начальных данных (скоростей и координат частиц) получить их траектории в любой необходимый последующий момент времени. Существует достаточно много различных методов интегрирования уравнений движения. Все они основаны на методе конечных разностей, где время изменяется дискретно с некоторым шагом Δt.
Среди наиболее известных методов интегрирования уравнений движения можно выделить алгоритм Верле [102], алгоритм «прыжков лягушки» (leap frog) [103], метод предиктора-корректора [104] (рис. 1-14). В данной книге будет описан первый из этих алгоритмов.
Алгоритм Верле в литературе по численному анализу часто называется неявной симметричной разностной схемой.
Основная идея алгоритма Верле состоит в записи разложения
положения частицы
и
:
|
|
|
Складывая (1.200) и (1.201), можно получить
|
где
ускорение
частицы (
), которое, однако, можно вывести также из
выражения
|
(1.203) |
Выражение (1.202)
это
основное уравнения алгоритма Верле. Данный алгоритм легко использовать, он
достаточно точен и стабилен, что объясняет его большую популярность в
МД-расчетах. Некоторым его недостатком является то, что он
не
самостартующий и необходимо использовать другой алгоритм для получения
нескольких первых точек координат.
Кроме вышеописанной версии алгоритма Верле, разработан скоростной алгоритм Верле, где положения, скорости и ускорения на шаге t + Δt вычисляются следующим образом:
|
Преимуществом скоростной формы алгоритма Верле является то, что она является самостартующей.
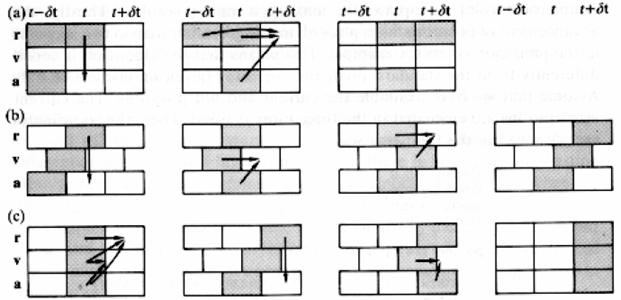
Рис. 1-14. Схематическое
описание различных форм алгоритма Верле: а
основной
алгоритм Верле (1.202); b
алгоритм
«прыжков лягушки»; c
скоростная
форма алгоритма Верле (1.204)
[105].
1.3.3.2 Неэмпирическая молекулярная динамика
Метод неэмпирической молекулярной динамики, впервые
предложенный в работе Кара
Паринелло
(Car
Parinello) [106], имеет широкое применение в современной физике и
химии. В нем потенциальная энергия системы, необходимая для нахождения сил,
действующих на атомы, выбирается не параметрически, а рассчитывается
квантово-химическим способом для любой конфигурации системы в ходе
компьютерного моделирования. В подходе неэмпирической молекулярной динамики
электронная система описывается набором волновых функций
,
которые принадлежат (или лежат очень близко к) основному состоянию для
потенциальной поверхности Борна
Оппенгеймера
в любой момент времени, что позволяет отразить совместное движение электронов и
ядер, описываемых набором координат
.
При этом поведение системы строится исходя из лагранжиана системы, где вводятся
фиктивное время и фиктивная масса электронов μ. В предположении, что величина
μ >> me,
фиктивная кинетическая энергия электронов будет оставаться малой по сравнению с
кинетической энергией ионов, что позволяет рассчитывать силы, действующие на
ядра в любой момент времени, по теореме Гельмана
Фейнмана
для электронных систем, соответствующих мгновенным ядерным конфигурациям
(адиабатическое приближение). Уравнения движения полной динамической системы,
включающей фиктивную электронную динамику и действительную ионную динамику,
определяются лагранжианом
|
(1.205) |
где
функционал
полной энергии, получаемый в любом квантово-химическом методе, набор {αν}
любые
возможные внешние параметры
температура,
давление, объем и пр., μ
фиктивная
масса или параметр инерции, связанный с орбитальными степенями свободы, μν
произвольный
параметр соответствующей размерности. Матрица
является набором множителей Лагранжа, которые
обеспечивают ортонормированность
.
Из этих уравнений можно получить уравнения движения Эйлера
Лагранжа
[107]:
|
(1.206) |
|
(1.207) |
|
(1.208) |
Данная вычислительная схема широко применялась, в частности, для моделирования отжига (simulated annealing), в ходе которого температура системы медленно понижалась, пока система не застывала в одном из самых глубоких локальных минимумов полной энергии. Тем самым решалась задача нахождения минимума энергии системы, что является очень сложной задачей для стандартных алгоритмов оптимизации.
Впоследствии данная схема молекулярной динамики была модифицирована, и сейчас наиболее широко применяются схемы без введения фиктивного времени и фиктивной массы электронов. Это позволяет прослеживать динамику системы в реальном времени, правда, за это приходится платить нахождением точных (или почти точных) волновых функций и уровней электронов при каждой вариации координат атомов. Но с развитием итерационных алгоритмов диагонализации больших матриц это уже не является столь трудной задачей, как раньше [26].